Introduction
Chemical modification of polymer surfaces is important in a wide variety of practical industrial technologies [1-6]. Particularly gaseous active species excited by gas molecules have been widely used to modify the surface of various polymers. These modification methods are available in corona, flame, and plasma treatments and are convenient techniques for surface modification of polymers. For example, under plasma oxidation processes, functional groups can be introduced at polymer surface layers to increase polar components in its free energy [4]. The gaseous treatments on polymer surfaces require the impingement of reactive species such as oxygen radicals [6, 9], which are useful and effective for such treatments. In recent years, green chemistry techniques have attracted much interest to modify surfaces under mild and safe conditions without high-energy consumption. Namely, using active species in gaseous phase is needed to be developed as a new technology. Thus, we have focused on O3, which has potential ability to modify polymer surfaces. Comparing to the discharged plasma technique, O3 treatment has some advantages; this system provides environmental safe species, since O3 only decomposes to O2 [7, 8]. In addition, polymer surfaces are modified under atmospheric pressure with O3. But the treatment efficiency of only O3 seems to be much lower than that of plasma treatment. So, recent examples for O3 treatment include ultraviolet light (UV) O3 exposure to polymer surfaces [9, 10]. In this case, UV light decomposes O3 to form active species and then treatment proceeds [7, 10, 11]. However, little work has been reported in O3 treatment for polymer surface modification. In the present work, polymer surface modification by thermal-O3 treatment of polymer is described. Since O3 is thermally decomposed to O2 and O [8], the O3 process is expected to achieve high efficient modification for polymer similar to UV/O3 treatment. After the thermal-O3 treatment, the surface properties of polyethylene (PE), poly (vinylchoride) (PVC), and polystyrene (PS) (Scheme 1) were evaluated by using DRS-FT-IR, UV/VIS/NIR spectroscopy and contact angle measurement. Especially, temperature dependence of polymer-O3 modification was focused.
Scheme 1. Chemical structure of polymers.
Experimental
Materials and Sample Preparation
Sample preparation of polymer for O3 treatment was carried out as follows: Polyethylene (PE) (Merck, Germany, MW = 66000), Poly(vinylchoride) (PVC) (Nipon ZEON, Japan, Mw=65000) and polystyrene (PS) (Nacalai tesque, Japan, Mw=120000) were used as raw polymers. When PE film was formed, PE powder was dried for 24 h at 60°C in vacuum and was heated at 150°C on a glass plate. The molten PE formed a film with about 150 µm thickness. In PVC and PS, wet cast procedure was utilized; 5 wt% concentration of polymer was dissolved in 50 ml tetrahydrofran (THF) for 24 h at 40°C. Each polymer solution was cast at 25°C on a glass plate (200 x 200 mm2) with about 100 µm thickness and then, coagulated immediately in water at 25°C. After solidification the films were washed well with distilled water to remove THF solvent and dried at 40°C in vacuo for 24 h. The water used in all experiments was prepared in purification system (Yamato, Japan, WG221).
Ozone Generation and Polymer Treatment
For polymer modification, the prepared polymer sample was set into the bottom of a pyrex glass cylinder (20 mm outer diameter and 200 mm in height) as illustrated in Figure 1. The polymer in the cylinder was heated by an oil bath in the range of 25-65°C. O3 was generated with oxygen gas by silent discharge ozonizer (ZC60-MM, Silver Seiko Ltd., Japan). At 780 Torr (1 Torr = 133 Pa), O2 (99.9%, Taiyo Sanso, Japan) was introduced to the ozonizer as O3 source. The flow rate of O2 (1.0 Nl/min) was controlled by a flowmeter (Kusano, FT-1100) and the total pressure of O2 flow was measured by a Baratron pressure 122 A. To generate O3 with 3026 ppm (w/v) concentration (1.35x10-4 mol/l), the four ozonizer units were connected in series (Figure 1). The control of O3 concentration was performed using a transformer, which charged electric voltage with 140 V. O3 concentration was measured upstream on the cylinder equipment by using UV spectrophotometer (Shimadzu UV-190) to monitor the maximum of O3 band at 254 nm Hartley [12].
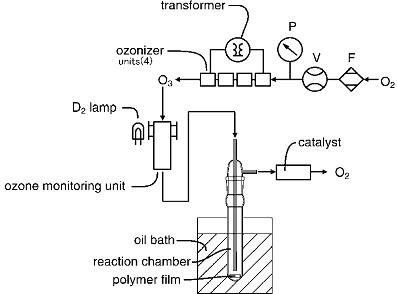
Figure 1. Schematic diagram of O3 flow system for polymer treatment. To evaluate O3 concentration UV spectroscopic instrument is included in silent discharged line. P: pressure gauge, V: valve, F: flow meter.
Ozonolysis of polymer was carried out in the range of 25°C and 65°C under O3 exposure for various treatment times. After O3 treatment of polymer, FT-IR spectroscopy (Shimadzu FTIR 8100) was used to evaluate the treated surface by diffuse reflectance spectroscopy (DRS) (Shimadzu DRS-8000) method. The IR spectra of the treated polymers surface were taken with a resolution of 4 cm-1 in spectral range from 450 to 4000 cm-1. For one spectrum 40 scans were averaged. The treated polymer surface was also characterized by measuring contact angle of pure water. The values of contact angles were measured using TYPE CA-A (Kyouwa Chemical Industry Co. Ltd). The reflection and absorption spectra of the treated surfaces in ultraviolet-visible-near infra-red (UV-VIS-NIR) region were measured with a Jasco V-570 spectrometer. In the region of UV-VIS-NIR, reflected beam from sample surface was collected by integration sphere to PbS detector (Jasco ISN-470).
Results and Discussion
FT-IR Analyses of Ozonized Polymers
To observe polymer surfaces treated with O3 at different temperatures, DRS-FT-IR spectra were taken after O3 treatment. Figure 2 shows FT-IR spectra of raw PE (a) and treated PE by O3 for 3 hour at 25(b), 45(c) and 65°C (d). The spectrum (a) shows three characteristic IR bands at 720, 1481 and 2927 cm-1, which were assigned to CH2 rocking, CH2 scissoring, and CH2 stretching, respectively [13, 14]. We noted that the FT-IR profile of spectrum (b) was very similar to that of (a) for untreated PE, but a new peak at 1720 cm-1 appeared by O3 exposure in (b). When the treatment temperature increased to 45°C and 65°C, IR bands appeared near 1100 and 1720 cm-1, which were attributed to C-O and C=O stretching, respectively. These IR intensities gradually increased by increasing the O3 exposure temperature. We confirmed that these bands were not observed at 65°C for 6 h without O3. Also, similar spectrum with Figure 2 (b) was observed at 25°C for 6 h with O3. Therefore, there was tendency that the formation of the C-O and C=O groups on PE under O3 atmosphere increased by increasing temperature.
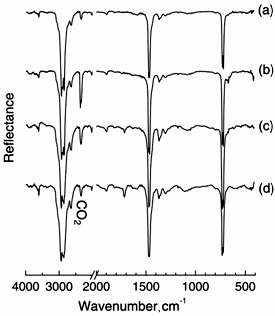
Figure 2. FT-IR spectra of PE (a) exposed to ozone at 25 (b), 45 (c) and 65°C (d) for 3 hours.
DRS-FT-IR Spectra of PVC
Similarly, DRS-FT-IR spectra of PVC were also measured after O3 exposure for 3 h at various temperatures. In Figure 3, the untreated PVC (a) showed characteristic IR bands at 617 and 693 cm-1 for C-Cl stretching, 1440 cm-1 for C-H bending, and 1260, 1325, and 2964 cm-1 for C-H stretching. The appearance of C=O band at 1724 cm-1 means that the surface of untreated PVC contained few C=O groups by oxidization. As well as PE, the spectra of PVC exposed at 25, 45 and 65°C were measured. However, a significant difference in the IR spectra of PVC after treatment was not observed in these cases. Figure 4 shows the FT-IR spectra of PS measured after the film was treated with O3 for 3 h at various temperatures. In spectrum (a) of untreated PS, characteristic IR bands of aromatic C-H stretching at 3010 and 3080 cm-1 and overtone and combination band due to C-H out-of-plane at 1600-2000 cm-1 were observed. Also, IR band of aromatic ring CC stretching and aromatic C-H in-plane appeared in 1000-1200 cm-1 region. When the PS film was treated with O3 at 25°C, resultant IR spectrum remarkably changed at 1743 cm-1 for C=O stretching. In spectra (c) and (d) for PS treated at 45°C and 65°C with O3, the 1743 cm-1 band was also observed. We noted that appearance of the stretching band of C=O was independent of exposure temperature.
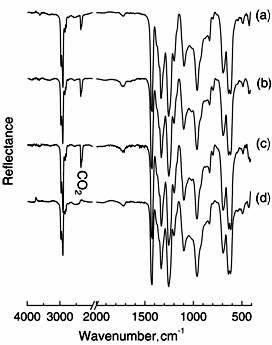
Figure 3. FT-IR spectra of PVC (a) exposed to O3 at 25 (b), 45 (c) and 65°C (d) for 3 hours.
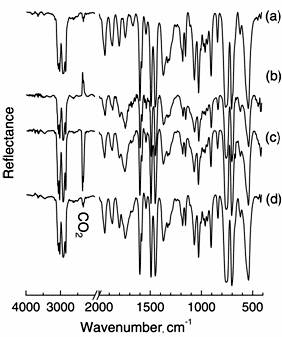
Figure 4. FT-IR spectra of PS (a) exposed to ozone at 25 (b), 45 (c) and 65°C (d).
Effect of Exposed Temperature on IR Spectra
To summarize effect of exposed temperature on IR spectra of PE, PVC and PS, ratio of each IR band intensity to CH2 stretching band near 2927 - 2970 cm-1 was compared. The resultant ratio of the IR bands was plotted with temperature. Figure 5 shows change of the values of each ratio observed at 25, 45 and 65°C. The left side points in each figure were IR data obtained without O3 exposure at 25°C. As temperature increased in PE from 25°C to 65°C, the ratio of C=O at 1720 cm-1 to CH2 stretching at 2927 cm-1 gradually increased from 0.02 to 0.18, respectively. But the values of PVC for C=O against the CH2 stretch 1724 cm-1/2964 cm-1 show little changes compared to the untreated data. In PS a C=O band at 1743 cm-1 significantly appeared as shown in Figure 4. The values of the ratio of the 1743 cm-1/ 2970 cm-1 bands were 0.7 and 0.8 at 25°C and 65°C respectively. Then, under O3 exposure, a little change of their values of the ratio was observed in the range of 25°C to 65°C. In the values of the ratio of 1050 cm-1 to 2970 cm-1 for phenyl ring CC stretching to the CH2 stretching, there was tendency to reduce of the phenyl ring band when the temperature became high. It was clear that decrease of the phenyl ring band corresponded to the increase of the C=O band in PS, as the temperature became high from 25°C to 65°C under the O3 exposure. When temperature increased in the presence of O3, it is known that self-decomposition of O3 proceeded [8]; O3 → O2 + O. The generated O species improved the formation of the C=O group on the PE surface. Figure 6 presents ratio of the IR intensity observed on polymers treated at various exposure times at 65°C. When the exposure time increased, little change was in the C=O band intensity for PVC. However, the C-Cl at 617 cm-1 was increased as compared with that of the CH2 stretching at 2964 cm-1 in O3. In PS, the value of C=O band increased with increasing the treatment time up to 30 min and then became constant around the value of 0.7~0.8. In addition, the data indicated that the phenyl ring band at 1050 cm-1 was reduced by the O3 exposure. In contrast to PS, a little change in the value of the ratio of 720 cm-1 to 2927 cm-1 was observed for the treated PE. It was reasonable to consider that the formation mechanism of C=O was different in PE and PS. Furthermore, it was very interesting to note that the formation of C=O group on PE surface had induction period [15] in the range of 0 to 30 min.
The result of PE indicated that temperature depending step was to be rate determining of the C=O formation on the PE surface. Therefore, it was reasonable to consider that O3 decomposition was not enough to treat the surface within 30 min. Namely, longer treatment time enhanced the O3 decomposition to O species near the polymer. The resultant O attacked the PE effectively to form the C=O group on the surface. Although the change was little in IR data of PVC, similar tendency was obtained with those of PE. Since oxidation process of PE depended upon exposure temperature, O3 decomposition on PE surface generated active species such as hydroxyl radical ( ) [7, 16, 17]. On the other hand, no induction period was observed in PS. In addition, the C=O formation on PS surface was independent of exposure temperature. These suggested that the PS surface was treated with O3 via ozonized intermediate species [10], which were formed directly by thermal decomposition of interaction between O3 and phenyl ring. As well known in O3 reaction with unsaturated carbon bond such -C=C- group [10, 18-21], it was reasonable for the O3-PS to consider ozonide formation between the phenyl ring and O3. Therefore, the C=O group maybe appeared after the ozonide complex formed; decomposition of the ozonide was the reason for C=O formation on the PS surface. ) [7, 16, 17]. On the other hand, no induction period was observed in PS. In addition, the C=O formation on PS surface was independent of exposure temperature. These suggested that the PS surface was treated with O3 via ozonized intermediate species [10], which were formed directly by thermal decomposition of interaction between O3 and phenyl ring. As well known in O3 reaction with unsaturated carbon bond such -C=C- group [10, 18-21], it was reasonable for the O3-PS to consider ozonide formation between the phenyl ring and O3. Therefore, the C=O group maybe appeared after the ozonide complex formed; decomposition of the ozonide was the reason for C=O formation on the PS surface.
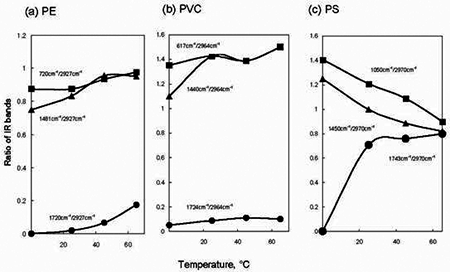
Figure 5. Plots of ratio of IR bands versus treated temperature for O3 exposure on (a) PE, (b) PVC and (c) PS. Description of the value of IR bands was indicated in each figure.
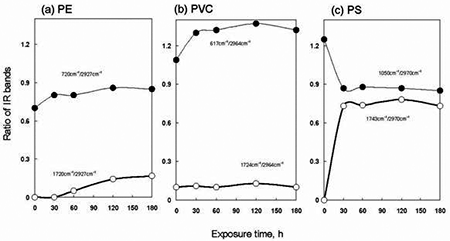
Figure 6. Plots of ratio of IR bands observed in (a) PE, (b) PVC and (c) PS in O3 in treated for different exposure times. The temperature was 65°C.
Reflectance Absorption Spectra
As shown in Figure 7, reflectance absorption spectra of PS supported introduction of the C=O group to the PS. That is, the absorption band of 220 nm region due to C=O was increased and band became broaden near 268 nm [13]. This supported that, obviously, aromatic rings of PS was modified with O3. For PE (traces (a)), the CH2 peak at 1731 nm was observed, but, changed after the O3 treatment. In addition, the absorption band of 200-250 nm region for PE significantly increased by the O3 treatment. In the UV region, it was reasonable to consider that hydrogen atom of PE was subtracted from PE surface and then oxygen species was introduced as a form of C=O group. However, PVC spectra presented few change of the absorption band. This suggested that chloride atom might interfere such O3 attack on polymer surface.
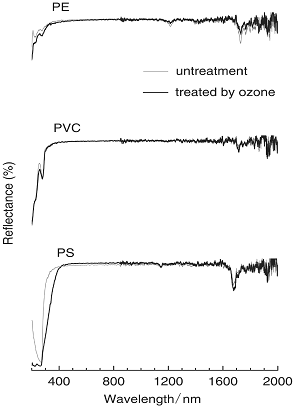
Figure 7. Reflectance absorption on spectra of PE, PVC and PS without and with O3 treatment at 65°C for 3 hours.
Effects of Ozone Treatment
In order to evaluate the effects of O3 treatment at 65°C, contact angle measurements were performed as shown in Figure 8. The values of contact angles of pure water were measured at 20°C on PE, PVC, and PS films for each polymer surface treated at 65°C in the presence of O3. Also, for reference data the polymers were treated with O3 gas at the temperature with different exposure times. In the case of PVC (b), little change was observed in the contact angle, even though increasing of the treatment time was kept for 3 h. For PE (a), the value of contact angle decreased with increase of O3 treatment time for 1 h and then became almost constant up to 3 h at about 81.2°. Figure 9 lists values of contact angles of water for these polymer surfaces, treated with O3 at 25°C and 65°C. It was observed in PE that resultant value of contact angle observed at 65°C was higher than those at 25°C. Namely, the value of 102.7 ° for the 25°C treatment was larger than that of 81.2° for 65°C. It was clear that O3 treatment at higher temperature enhanced the degree of hydrophilic nature of PE. In contrast, plots (c) in Figure 8 showed that the values of contact angle decreased from 75.8° to 18° with increase of the exposure time. This suggested that the O3 exposure at 65°C improved surface wettability of PS. In addition, the hydrophilic nature of PS was remarkably increased relative to that of PE and PVC. As shown in Figure 9, the value of PS was 26.0° when the PS surface was treated by O3 at 25°C. The surface of PS was improved to be hydrophilic nature by only O3 exposure at the low temperature. In the O3-PS system, these facts well correspond to the experimental results of temperature dependence of C=O formation on FT-IR spectra. Similar result was also shown in the case of the O3-PE system. It is interesting to note the temperature dependence on the surface modification by O3 for PS and PE. In comparing contact angles measured at 25°C and 65°C, apparently, the sample treated at 65°C was somewhat higher than at 25°C. The increment of hydrophilic nature at 65°C was effective for formation of C=O group. This was due to effect of thermal-O3 treatment for PE. Relative to PE, PS modification mostly proceeded to be independent of the exposure temperature. Therefore, formation of C=O group improved surface wettability of PS was not due to mainly such thermal-O3 treatment; that is, in PS-O3 complex decomposed O3 under low temperature condition.
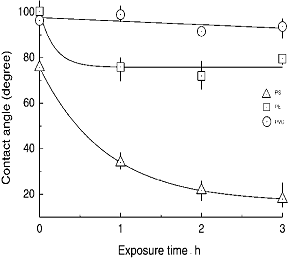
Figure 8. Plots of water contact angles against O3 exposed time for each polymer. O3 treatment was carried out at 65°C.
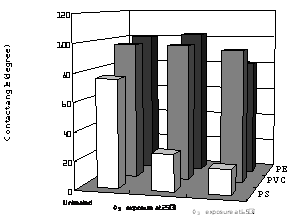
Figure 9. Water contact angles measured on each polymer surface. O3 exposure was carried out at 25°C and 65°C.
Conclusions
The present work focused on polymer modification by thermal O3 treatment. Surface characterization of the polymers was carried out using DRS-FT-IR and UV/VIS/NIR spectroscopy and contact angle measurements for treated PE, PVC, and PS. The results of DRS-FTIR spectra of polymers suggested that the C=O groups on the surface were formed on PE and PS treated at 65°C. For the C=O appearance there was tendency that the C=O band intensity of the PE was smaller than that of PS. It was found that the treatment of O3 on PS surface was independent of the exposure temperature, but in PE-O3. The surface oxidized and formed C=O group on the surface strongly depended on the exposure temperature. Therefore, we concluded that thermal-O3 treatments improved the PE surface having wettability. On the other hand, in PS-O3, O3 directly oxidized the PS surface regardless of exposure temperature. This caused that O3 treatment enhanced the wettability of the PS surface without higher temperature.
Acknowledgements
This work was partially supported by the 21st century COE program, creation of hybridized materials with super functions and formation of an international research education center of Nagaoka University of Technology.
References
1. M. J. Wang, Y. I. Chang and F. P. Epaillard, " Effects of the Addition of Hydrogen in the Nitrogen Cold Plasma: The Surface Modification of Polystyrene", Langmuir, 19, 8325-8330 (2003).
2. M. P. Mendoza, M. D. Garcia and F. J. L. Garzon, "Modifications Produced by O2 and CO2 Plasma Treatments on a Glassy Carbon: Comparison with Molecular Gases", Carbon, 37, 1463-1474 (1999).
3. C. Seidel, H. Kopf, B. Gotsmann, T. Vieth, H. Fuchs and K. Reihs, " Ar plasma Treated and Al Metallised Polycarbonate: a XPS, Mass spectroscopy and SFM study", Applied Surface Science, 150, 19-33 (1999).
4. M. J. Shenton and G. C. Stevens, "Surface Modification of Polymer Surfaces: Atmospheric Plasma versus Vacuum Plasma Treatments", J. Phys. D: Appl. Phys., 34, 2761-2768 (2001).
5. E. F. Abdrashitov and A. N. Ponomarev, "Plasma Modification of Elastomers", High Energy Chemistry, 37, 279-285 (2003).
6. H. Kumagai, H. Denbo, N. Fujii and T. Kobayashi, "Poly (ethylene terephthalate) Decomposition Process in Oxygen Plasma; Emission Spectroscopic and Surface Analysis for Oxygen-plasma Reaction", Journal Vacuum Science Technology A, 22, 1-7 (2004).
7. H. N. Lim, H. Choi, T. M. Hwang and J. W. Kang, "Characterization of Ozone Decomposition in a Soil Slurry: Kinetics and Mechanism", Water Research, 36, 219-229 (2002).
8. S. Migita, K. Sakai, Z. Mori, H. Ota and R. Aoki, "Evaluation of Ozone Condensation System by Thermal Decomposition Method", Jpn. J. Appl. Phys., 36, 94-97 (1997). Japanese Journal of Applied physics.
9. K. Efimenko, W. E. Wallace and J. Genzer, "Surface Modification of Sylgard-184 Poly(dimethyl siloxane) Networks by Ultraviolet and Ultraviolet/Ozone Treatment", Journal of Colloid and Interface Science , 254, 306-315 (2002).
10. T. N. Murakami, Y. Fukushima, Y. Hirano, Y, Tokuoka, M. Takahashi and N. Kawashima, "Surface Modification of Polystyrene and Poly(methyl Methacrylate) by Active Oxygen Treatment", Colloids and Surfaces B, 00, 1-9 (2002).
11. I. Mathieson and R. H. Bradley, "Improved Adhesion to Polymers by UV/ozone Surface Oxidation", International Journal. Adhesion and Adhesives, 16, 29-31 (1996).
12. R. W. B. Pearse and A. G. Gaydon, "Iden OF Molecular Spectra", Chapman & Hall LTD., London (1950).
13. R. M. Silverstein and F. X. Webster, "Spectrometric Identification of Organic Compounds", Wiley, N.Y. (1998).
14. G. Socrates, "Infrared Characteristic Group Frequencies", Wiley, N.Y. (1980).
15. S. A. Carr and R. B. Baird, " Mineralization as a Mechanism for Toc Removal: Study of Ozone/ozone-Peroxide oxidation using FT-IR", Water Research, 34, 4036-4048 (2000).
16. E. J. Feltham, M. J. Almond, G. Marston and V. P. Ly, "Reaction of Alkenes with Ozone in the Gas Phase: a Matrix-isolation Study of Secondary Ozonides and Carbonyl-containing Reaction Products", Spectrochimica Acta A, 56, 2605-2616 (2000).
17. Q. K. Timerghazin, S. L. Khursan and V. V. shereshovets, "Theoretical Study of the Reaction between Ozone and C-H bond: gas-phase reactions of hydrocarbons with ozone", Journal of Molecular Structure, 489, 87-93 (1999).
18. S. Toby and F. S. Toby, "Reactivity of the Ozone-Ethane system", Journal of physical chemistry A, 102, 4527-4531 (1998).
19. O. V. Manoilova, J. C. Lavalley, N. M. Tsyganenko and A. A. Tsyganenko, "Low-temperature IR Study of Ozone Interaction with Etthylene Adsorbed on Silica", Langmuir, 14, 5813-5820 (1998).
20. D. Heymann, S. M. Bachilo, R. B. Weisman, F. Cataldo, R. H. Fokkens, N. M. M. Nibbering, R. D. Vis and L. P. F. Chibante, "C60O3, a Fullerene Ozonide: Synthesis and Dissociation to C60O and O2 ", Journal of American Chemical Society, 12, 11473-11479 (2000).
21. D. B. Mawhinney and J. T. Yates Jr., "FTIR Study of the Oxidation of Amorphous Carbon by Ozone at 300 K - Direct COOH formation", Carbon, 39, 1167-1173 (2001).
Contact Details
|