Introduction Before 1970, the nuclear power industry strategy for high-level radioactive waste (HLW) was to immobilise it in borosilicate glass, followed by deep burial in the Earth. However in the early 1970s, geochemists realised that borosilicate glasses might not be particularly stable with respect to contact by groundwater when buried in the ground, because a) radiogenic heating within the glass and/or the geothermal gradient could produce temperatures well in excess of 100°C, and b) groundwater could not be guaranteed to avoid contact with the glasses, even if a series of additional barriers such as metal containers and clay overpacks were introduced into a geological repository. Alternative disposal matrices (wasteforms) were first put forward for HLW at the Pennsylvania State University, with the basic idea of incorporating the waste fission products and associated actinides in the crystalline lattices of synthetic minerals, which were known to be very long-lived (many millions of years) in nature [1]. These minerals included silicates (pollucite, CsAlSi2O6; Sr-feldspar, SrAl2Si2O8), phosphates (monazite, CePO4; apatite, Ca5(PO4)3(F,OH,Cl)) and oxides (fluorite-structured UO2). Mixtures (assemblages) of these phases were formed by adding calcia, phosphate, alumina and silica to the fission product wastes and sintering at ~ 1100°C. Waste loadings in these “Supercalcines” were as high as 70 mass%. In 1979, Ringwood et al. [2] suggested assemblages of titanate minerals could be used to incorporate HLW, on the basis that in Nature, titanates are much more water-resistant than the supercalcine suite of minerals. In titanate assemblages waste ions are only dilutely incorporated into the phases, whereas in the supercalcines fission products/actinides were the basis of the phases. Because of the resemblance of the titanate phases to naturally occurring rocks, the alternate material was called SYNROC (synthetic-rock) Several forms of SYNROC have been named but the most extensively studied is SYNROC-C which was designed to incorporate Purex-type reprocessing waste from commercial nuclear power production [3]. SYNROC-C consists of four main titanate phases, ziconolite (CaZrTi2O7), “hollandite” (Ba(Al,Ti)2Ti6O16), perovskite (CaTiO3), titanium oxides (TinO2n-1) and ~ 2-5% metallic alloys. The first three phases provide various lattice sites in which nearly all the HLW ions are incorporated in solid solution. Titanium oxides are important - although they do not incorporate waste elements, they provide a chemical buffer that enables the relative proportion of SYNROC phases to vary in an in-situ manner. Some of these phases and their stability and the incorporation mechanisms of waste ions are discussed. Also another naturally occurring mineral, apatite is discussed with respect to immobilising niche waste 129I. Hollandite The hollandite structure may be stabilised with a diverse range of elements that conform to AxByC8-yO16, stoichiometry (x<2, y~1)[4]. The A site consists of large monovalent or divalent cations (Ba2+, Cs+, Rb+, etc) whilst the B and C sites may consist of various ions that exhibit valences between 2+ and 5+, typically transition metals. B cations are normally less abundant and have a lower valence than C-site cations. Hollandite is the host phase for Cs in the disposal matrix and typically displays BaxCsyAl2x+yTi8-2x-yO16 stoichiometry (1.08 < x+y < 1.51) [5]. Perovskite Perovskite, CaTiO3, is capable of incorporating a wide range of elements including sodium, strontium, rare-earths and actinides although it exhibits a strong preference for the larger (trivalent) ions compared to zirconolite. The structures of ABO3 perovskites consist of corner linked BO6 octahedra, which form a three-dimensional framework with large central cavities. The A cation is centrally located in these cavities in twelvefold coordination to oxygen. The perovskite structure is known to be stabilised in ABO3 compounds when the A cation is considerably larger than the B cation, specifically when the value of  is between 0.73 and 1.0, where r = ionic radius [6]. CaTiO3 exhibits orthorhombic symmetry at room temperature. is between 0.73 and 1.0, where r = ionic radius [6]. CaTiO3 exhibits orthorhombic symmetry at room temperature. Zirconolite Zirconolite is a key phase capable of incorporating actinide, or trans-actinium, elements (such as U, Np and Pu). It has a layered structure that exhibits a range of stoichiometries and polytypes, described by the general formula CaZrxTi3-xO7. The zirconolite polytypes are characterised by the number of layers that constitute a repeat and the manner in which they are stacked. All the zirconolite polytypes are derived from an anion-deficient fluorite superstructure [7, 8]. The polytype most commonly referred to as zirconolite is zirconolite-2M. This is monoclinic, has a two layer repeat and displays CaZrxTi3-xO7 stoichiometry for 0.80<x<1.37, see Figure 1. The Ca site in zirconolite-2M is eight coordinated, the Zr-site is seven coordinated and there are three distinct Ti sites, two of which are six-coordinated and the third, half-occupied site, five coordinated. Rare-earth and actinide elements may be incorporated on either the Ca or Zr sites, whilst transition metal corrosion products may be substituted on the Ti sites. Both Kesson et al. [9] and Vance and Agrawal [10] showed that the progressive replacement of Zr in zirconolite by U led to the formation of a pyrochlore structure when the replacement exceeded ~ 0.5 formula units (fu), with the pyrochlore phase being present for U replacements up to 1 fu. A more detailed analysis was published recently [11]. | 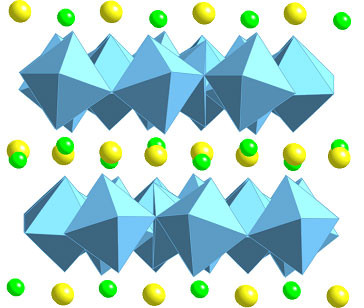
| | Figure 1. The (110) structure of zirconolite-2M. The interlayer Ca (large) and Zr (small) cations are shown between the planar octahedral Ti layers. | | 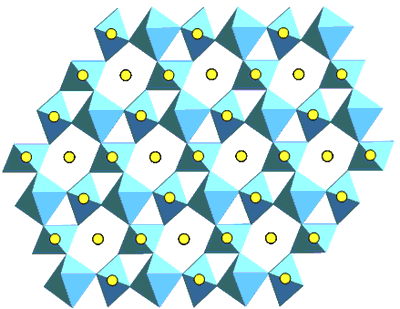
| | Figure 2. The (110) structure of pyrochlore showing the planar arrays of corner linked TiO6 octahedra and the A-site cations. | Pyrochlore Pyrochlore is a cubic structure which may be represented as A2B2O7 where A and B correspond to trivalent and tetravalent sites (or divalent and pentavalent) respectively. The A-site is eight coordinate and the B-site six-coordinate. Pyrochlore is able to incorporate rare earths and actinides in greater concentrations than zirconolite. Hf and Gd are two neutron absorbers which can be readily incorporated in these phases to facilitate high fissile actinide concentrations with criticality control. Zirconolite – Pyrochlore System The zirconolite-pyrochlore titanate system spans a large matrix of single oxides, viz. CaO-ZrO2-TiO2-AnO2-An2O3-M2O3-MO2 etc., where An = actinide ions and the metal (M) ions are appropriate charge compensators. Vance et al. [12] have shown for compositions CaZr1-yPuyTi2O7 with y up to 0.5 fu, a zirconolite structure still survived, but at y = 0.8 fu the structure was pyrochlore. It was more recently shown [13] that for U and Pu substitution in the eight-fold site of pyrochlore, the U plus Pu inventory required must exceed ~ 0.6 fu. (at smaller U and Pu contents, zirconolite-2M and -4M polytypes are formed) notwithstanding the presence of 0.22 fu trivalent rare earths (Gd and Sm) [14]. If Zr in zirconolite is replaced by Hf and the A-site in pyrochlore by Gd, then the structures CaHfTi2O7 and Gd2Ti2O7 are formed. This system is targeted for Pu immobilisation and Gd and Hf are neutron absorbers as discussed above. Beginning with CaHfTi2O7, the progressive replacement of Ca2+ and Hf4+ ions by two Gd3+ ions results in the phase changes shown in Figure 3. In this system almost pure zirconolite-4M is formed between x=0.43-0.72 in CaxGd2(2-x)HfxTi2O7 as shown in Figure 3 [15]. When x<0.83 only zirconolite-2M is present and both are present between x =0.72 to 0.83. In general between 0.5 and 0.7 Zr fu gives rise to either or both zirconolite polytypes depending on the impurity cations present [15]. The numerous polytypes of zirconolite have been enumerated by Smith and Lumpkin [16]. In some surplus Pu waste streams, a variety of cation and anion impurities were present [17]. The addition of divalent cation such as Mg2+ and trivalent cation Al3+ to this system where valence states of the impurity cations are fixed enabled to study the phase stability for the potential Pu waste steams [15]. When Mg2+ was added in the form of Mg2TiO5 the pyrochlore phase region was extended [15]. This system has the potential to incorporate Pu waste streams containing variety of cations and anion impurities. | 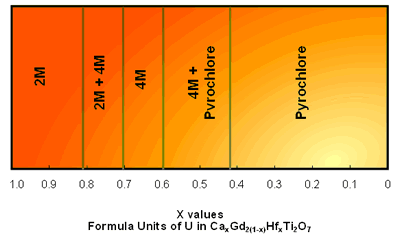
| | Figure 3. CaHfTi2O7 - Gd2Ti2O7 pseudo-binary phase system in air heated at 15000C for 96h. | Apatite Apatite (Ca5(PO4)3(F,OH,Cl)) is a naturally occurring mineral that accepts numerous substitutions of both cations and anions. Elements such as U and Sm and their isotopes are accommodated in nature [18]. One of its members is fluorapatite (Ca5(PO4)3F) which occurs naturally. It has a hexagonal structure and is the space group P63/m [18]. It was considered as a host matrix to incorporate 129I by hot isostatic pressing [19], but it was not possible because the ionic radii of F- is much smaller than that of I- (see Table 1). In the literature substitution of (PO4)3- by many other groups including (VO4)3- have been reviewed [21]. Sudharsan et al. [22] have synthesised stable Cd10(VO4)6I2. As suggested by Audubert et al. [23], replacement of Ca with Pb or Cd would make the halide site larger and encourage it to fit in the I-. They reacted the iodine which was present as PbI2 with Pb3(VO4)2 to form Pb10(VO4)6I2. Pb2+ and V5+ are larger than Ca2+ and P5+ respectively, therefore more space was created to fit the larger I- ion. Conclusions In this brief review we have shown using crystal analogues of naturally occurring minerals which have retained radioactive elements over millions of years provide flexibility to incorporate all HLW containing ions. Understanding of the crystal chemistry plays an important role in designing host matrices for various wasteforms. Crystal chemistry plays an important role in designing host matrices for various wasteforms. Crystal chemical flexibility of each of the host matrix phases is an essential quality of a wasteform. The crystal chemistry of zirconolite, pyrochlore and apatite has been discussed as examples of this flexibility. Table 1. Crystal radii and coordination number of cations and anions forming apatite from [20] | | | Ca2+ | 8 | 0.126 | | P4+ | 4 | 0.031 | | F- | 6 | 0.119 | | O2- | 6 | 0.124 | | Pb2+ | 6 | 0.133 | | V4+ | 4 | 0.050 | | I- | 6 | 0.206 | References 1. G. J. McCarthy, “High-level Waste Ceramics:Materials Considerations and Product Characterization”, Nucl. Tech., 32 (1977) 92. 2. A. E. Ringwood, E. S. Kesson, N. G. Ware, W. Hibberson and A. Major, “Geological Immobilisation of Nuclear Reactor Wastes,” Nature, 278 (1979) 219. 3. A. E. Ringwood, E. S. Kesson, K. D. Reeve, D. M. Levins and E. J. Ramm, “Synroc”, in “Radioactive Waste Forms for the Future”, ed. R. C. Ewing and W. Lutze, Elsevier, North-Holland, Amsterdam, Netherlands, (1988) pp. 233-334. 4. A. Bystrom and A. E. Bystrom, “The Positions of the Barium Atoms in Hollandite”, Acta Cryst., 4 (1950) 469. 5. S. E. Kesson and T. J. White, “[BaxCsy][(Ti, Al)3+2x+yTi4+8-2x-y]O16 Synroc-type Hollandites, II. Structural Chemistry”, Proc. R. Soc. Lond., A (1986) 295-319. 6. R. Roth, “Classification of Perovskite and Other ABO3-type Compounds”, J. Res. Natl. Bur. Stand, 58 (1957) 75-88. 7. H. J. Rossell, "Zirconolite-a Fluorite-related Superstructure" Nature, 283 (1980), 282-283 8. B. M. Gatehouse, I. E. Grey, R. J. Hill and H. J. Rossell, “Zirconolite, CaZrxTi3-xO7; Structure Refinements for Near-end Member Compositions with x=0.85 and 1.30”, Acta Crystallogr. Sect., B37 (1981) 306-312. 9. S. E. Kesson, W. J. Sinclair and A. E. Ringwood, "Solid Solution Limits in Synroc Zirconolite", Nucl. Chem. Waste Management, 4 (1983) 259-265. 10. E. R. Vance and D. K. Agrawal, "Incorporation of Radionuclides in Crystalline Titanates", Nucl. Chem. Waste Management, 3 (1982) 229-34. 11. E. R. Vance, G. R. Lumpkin, M. L. Carter, D. J. Cassidy, C. J. Ball, R. A. Day and B. D. Begg, “Incorporation of Uranium in Zirconolite”, J. Am. Ceram. Soc., 85 (2002) 1853-1859. 12. E. R. Vance, B. D. Begg, R. A Day and C. J. Ball, "Zirconolite-based Ceramics for Actinide Wastes", Scientific Basis for Nuclear Waste Management XVIII, ed. T. Murakami and R.C Ewing, Matls. Res. Soc. Symp. Proc., 353 (1995) 775-782. 13. E. R. Vance, M. W. A. Stewart, R. A. Day, K. P. Hart, M. J. Hambley and A. Brownscombe, “Pyrochlore-rich Titanate Ceramics for Incorporation of Plutonium, Uranium and Process Impurities”, ANSTO Materials Division Report No. R97m030, 18 Nov. 1997, Australian Nuclear science and Technology Organisation, Australia. 14. B. D. Begg, R. A. Day and A. Brownscombe, “Structural Effect of Pu Substitutions on the Zr-site of Zirconolite”, in “Scientific Basis for Nuclear Waste Management XXIV”, eds. K.P. Hart and G.R. Lumpkin, Matls Res. Soc. Sym. Proc., 663 (2001) 259-66. 15. D. S. Perera, M. W. A. Stewart, H. Li, A. Day and E. R. Vance, “Tentative Phase Relationships in the System CaHfTi2O7-Gd2Ti2O7 with up to 15 mol% additions of Al2TiO5 and MgTi2O5”, J. Am. Ceram. Soc., 85 (2002) 2912-2924. 16. K. L. Smith and G. R. Lumpkin, “Structural Features of Zirconolite, Hollandite and Perovskite, the Major Waste-Bearing Phases in Synroc”, in “Defects and Processes in the Solid State: Geoscience Applications”, The McLaren Volume, ed. J. N. Boland and J. D. Fitzgerald, Elsevier, Amsterdam, Netherlands, (1993) pp.401-22 . 17. M. W. A. Stewart, E. R. Vance, R. A. Day, S. Leung, A. Brownscombe, M. L. Carter and E. B. Ebbinghaus, “Impurity Incorporation in Pyrochlore-rich Ceramics”, Ceram. Trans., 107 (2000) 569-576. 18. J. M. Hughes and J. Rakovan, “The crystal structure of apatitite, Ca5(PO4)3(F,OH,Cl)”, in Reviews in Mineralogy & Geochemistry”, 48 (Phosphates, Mineralogical Society of America, ed. , M. J. Kohn, J. Rakovan and J. M. Hughes), Pub. Mineralogical Society of America, Washington DC, USA, (2002) pp. 1-12. 19. D. S. Perera, B. D. Begg, R. L. Trautman, D. J. Cassidy, E. R. Vance and R. A. Day, “Hot isostatic Pressing Method for Immobilising Radioactive Iodine-129”, in “International HIP Conference, HIP’02”, Moscow, Russia, May 20-22, 2002, All Russia Institute of Light Alloys, Moscow, (2003) 162-167. 20. R. D. Shanon, “Revised Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides”, Acta Cryst., A32 (1976) 751. 21. Y. Pan and M. E. Fleet, “Compositions of the Apatite-group Minerals: Substitution Mechanisms and Controlling Factors”, in ref 18, pp. 13-49. 22. K. Sudarsana, R. A. Young and A. J. C. Wilson, “The structure of Some Cadmium ‘Apatites’ Cd5(MO4)3X: I. Determination of the Structure of Cd5(VO4)3I, Cd5(PO4)3Br and Cd5(VO4)3Br”, Acta. Cryst., B33 (1977) 3136-3142. 23. F. Audubert,, J. Carpena, J. L. Lacout and F. Tetard, “Elaboration of an Iodine-Bearing Apatite: Iodine Diffusion into a Pb3(VO4)2 matrix”, Solid State Ionics, 95 (1997) 113-119. Contact Details |