Mar 3 2016
Perovskite has a hexagonal or rhombohedral structure with formula ABO3, while still belonging to the R3c space group (number #161). They possess ferroelectric, piezoelectric, and superconducting properties and are often used in applications of functional devices.
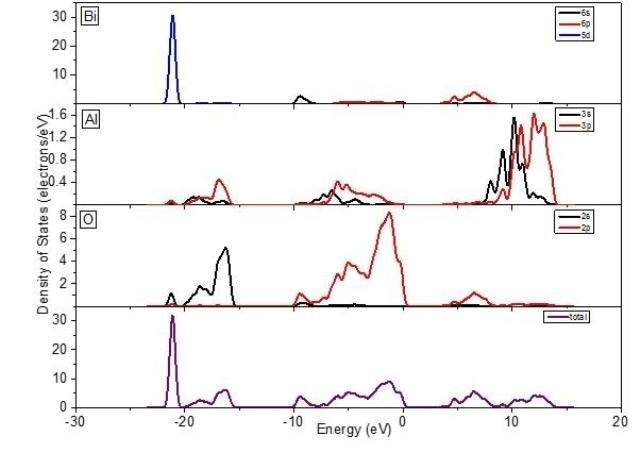 This is a diagram illustrating the variation of partial density of states for bismuth, aluminum and oxygen. credit: MPLB
This is a diagram illustrating the variation of partial density of states for bismuth, aluminum and oxygen. credit: MPLB
BiAlO3 was also used to enhance the electrical and structural properties of BNT-based ceramics, because of the huge polarization exhibited in the Perovskite structure. Perovskite is added into BaTiO3, PbZrO3, and PbTiO3-based ceramics to enhance their electrical properties. The optical and electronic properties in hexagonal and cubic have been analyzed by using first principle methods.
In this analysis, density functional theory (DFT) in tandem with local density approximation (LDA), is used to determine the vibrational, elastic, and electronic properties. The elastic quantities provided here are important to highlight structural and bonding stability, based on the examined anisotropic quantities.
In this study the researchers analyzed the crystal structure of perovskite BiAlO3 by initially using the calculations made by the DFT. They used the atomic positions provided by the previous literature to develop a lattice structure using the sophisticated ab initio DFT software.
An advanced structure like this is identified in the rhombohedral perovskite system with space group with R3c (#161) and lattice parameter of a = b = c = 5.338?A, bond angle of α = β = γ = 60?, while handling the exchange-correlation potential with the LDA technique. The calculations helped to identify the phonon, elastic, optical, and electronic properties.
The aspect covered in this study is considered to be a common research topic in perovskites ABO3. The readers of this work will be able to understand the essence of these vital topics and their corresponding developments in Material Physics and Engineering Chemistry.
The study was supported by the National Science Foundation of China (NSFC) project grant nos. 11234001 and 91433102, under the recipient of Prof. D. Yu.