There are many problems related to zeta potential represented by the Greek letter ‘ζ.’ Life-saving drugs, intended to appear clear prior to injection having large particles floating inside, paint caking, lumps in cooking sauces and cloudy tap water are just some such issues.
Zeta potential is linked to the charge density on small particlesi. In many, but not every case, the repulsive force that stops particles from aggregating is an outcome of this charge concentration. It cannot be measured directly; however, it may be understood via a determination of zeta potential.
The square of this potential is relative to the force of electrostatic repulsion. As this potential falls to zero, repulsion falls to zero and instability will ensue without steric repulsion. There are various methods to define zeta potential, but each one has a commonality: zeta potential is not directly measured; it’s determined from the electrophoretic mobility that is itself established from the electrophoretic velocity. Placing a charged particle in an electric field creates a movement known as electrophoresis.
The most renowned kind is gel electrophoresis, broadly employed in protein and DNA divisions. The medium is a sponge-like gel. A gel is not employed for particle or micro-electrophoresis; charged particles move in a liquid. It should be noted that when searching the internet for electrophoresis, it is likely that a lot more hits related to gel electrophoresis will occur. Searching instead for ‘zeta potential’ will result in more appropriate information. If there is a desire to purchase a zeta potential analyzer, the following general questions must be posed.
Classification of Various Techniques
- How to establish requirements: quantitative and qualitative?
- What methods are the most likely to solve the problem?
Classifying Techniques
Classification should begin by examining some frequently used ways to categorize commercially accessible methods. Methods can be categorized in one of three ways:
- Ensemble Averaging: Laser Light Scattering
- Concentrates: Acoustical Methods
- Imaging: Single Particle Observations
II.1 Classification: Imaging, Single Particle Observations
The classical method employed cells like those, which can be seen in Figure 1.
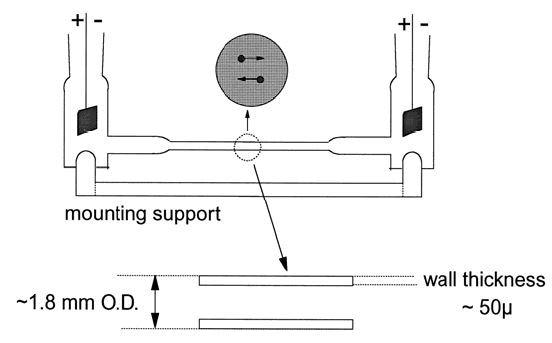
Figure 1. Van Gils’ classical micro-electrophoresis cell. Image Credit: Brookhaven Instrument Corporation
An applied voltage creates an electrical field, .png) Charged particles travel in the direction of the electrode of the opposite sign. Individual particles are timed using a stopwatch as they travel a distance established by the reticle in the microscope’s objective. The electrophoretic velocity, ܸ
Charged particles travel in the direction of the electrode of the opposite sign. Individual particles are timed using a stopwatch as they travel a distance established by the reticle in the microscope’s objective. The electrophoretic velocity, ܸ.jpg) is established. However, if the voltage is enhanced, the velocity is too. So that this is not a problem, an intrinsic property is specified, the electrophoretic mobility, µep. ܸ
is established. However, if the voltage is enhanced, the velocity is too. So that this is not a problem, an intrinsic property is specified, the electrophoretic mobility, µep. ܸ
 |
Equation 1 |
Despite this, due to the cell design, a counter flow occurs because of charged entities bonded to or adsorbed on the cell walls. This causes an intrusive electroosmotic velocity, Veo. If this is not prevented, the velocity noted is the total of electro-osmosis and electrophoresis.
Measurements need to be taken near to the cell walls, at what is known as the static planes, where the electro-osmosis cancels out. This condition is a restriction of these methods. Although the initial image analyzers for zeta potential studied one particle at a time, newer editions employ CCD cameras and automate the tracing of particles in the field. However, as single particle devices, they do not average many values to achieve average µep from which average ζ potential is determined.
There is a smaller threshold of particle size linked with any method where an image is necessary. Dependent on the optics, this restriction is somewhere close to 100 nm and lots of kinds of essential nanoparticles are precluded. There are varying relations between ζ potential and µep, but the most frequently occurring historic model is that of Smoluchowski as personified in the standard equation employed in all instruments:
 |
Equation 2 |
Where ε and η are the liquid’s comparative dielectric constant (permittivity) and bulk viscosity and εo is a constant.ii Usually, this equation only pertains to big particles (bigger than, extremely roughly, a few tens of nanometers) and large salt concentration (bigger than, extremely roughly, a few tens of millimolar). However, for historical purposes, if not otherwise stated, it is this connection that is employed to determine zeta potential.
II.2 Classification: Concentrates, Acoustic Methods
A 1% specification of 993 nm polystyrene latex suspended in the water looks like partially diluted milk. Apart from if the path length that is looked through is under a millimeter or two, the sample is too thick for projecting the image of particles moving in an electric field. It is too dense for any type of currently commercially accessible light scattering to ascertain zeta potential. The light is multiply scattered as, prior to its existence, it is expected to meet with and scatter off an additional particle. This is where acoustic methods can assist, from about 1% to 50% volume fraction.iii
It is relatively simple to dilute a 1% latex whilst maintaining the charge density near the particle. However, with a lot of industrial samples, that is not the case every time. If special precautions are not taken, the charge density alters. In certain instances, general emulsions, for instance, it could be challenging to maintain the charge density during dilution, but if measurements are taken soon after dilution, excellent results should be secured. In such instances, additionally, dilute with the equal concentration of dispersing agents that were in the initial sample—the same stands for microemulsions.
When an oscillating electric field is used on electrodes in a suspension of charged particles, whose concentration varies from that of the nearby medium, the particles oscillate, creating an extremely low sound wave. Finding this weak signal close to other effects (cross talk) needs a delay rod and a well-reasoned method to the applied frequency of the oscillating field. It can vary on many physical attributes of the particle and the liquid. This method is known as ESA, electrokinetic sonic amplitude.
A different method employs the sonic transducer to run the charged particles generating an alternating current that can be detected by electrodes close to the suspension. This is named CVI, colloid vibration current, or CVP, colloid vibration possibility.
Collectively these different methods -each possessing benefits and drawbacks reliant on the sample, its strength and the liquid- are known as EAA, electroacoustic attenuation. Either one or the other is the favored method if it is a necessary measure in concentrates. The mathematics that connects measured acoustic attributes (impedance, attenuation, speed) or electroacoustic property (vibration potential, current) and the connection to particle size distribution, charge density (leading to zeta potential) and concentration is complex.
Not possessing full comprehension of this is a flaw in the method, particularly for those who are inexperienced. Sizing may be carried out with acoustics and zeta potential with electroacoustic attenuation, but just for sizes small enough. How small is additionally dependent on particle density.
An additional obstacle is that size may additionally be acquired from electroacoustic attenuation by observing the frequency reliance of the phase. However, other particle and liquid attributes are participants.
II.3 Classification: Ensemble Averaging, Laser Light Scattering
The most frequently used zeta potential analyzers are founded on laser light scattering: either electrophoretic (ELS) or phase analysis (PALS). In ELS, a Doppler-shifted frequency is calculated from which the electrophoretic velocity can be determined. In PALS, a phase variation is calculated from which the electrophoretic velocity can be measured.

Figure II. Laser light scattering optical configuration for ζ potential determination. Image Credit: Brookhaven Instrument Corporation
In both instances, a small part of the initial beam (the reference beam) is re-routed and a static frequency conveyed such that once recombined with the scattered beam, it is the disparity that is noted. This permits the frequency and phase shift to be examined for positive and negative mobility and ζ potential.
This can be seen in Figure II. Prior to the insertion of the electrodes, if the reference beam gets obstructed and a digital autocorrelator is employed to evaluate the scattered light signal, this exact optical configuration may be employed as a dynamic light scattering particle sizer. (A photon counter is an element of a digital autocorrelator; for this reason, the strength of the scattered light may additionally be assessed.
This information additionally generates the molecular weight if the particles are dispersed proteins or polymers.) It should be noted that the classification of size and zeta potential are two distinct measurements, not symbiotic. Not as with the classical micro-electrophoresis instruments that are exploring at comparatively few particles, the number of particles in the light scattering volume is extremely big, typically a lot higher than several thousand. As such, these instruments are not single particle counters and the average outcomes are averaged across an extremely big amount of scattering particles.
However, to understand the scattering accurately, samples are dilute; if not, multiple scattering creates a mismatch across the internal scattering angle and where the detector is positioned. This agreement across many particles in the scattering volume but a dilute sample is simple to achieve with colloids, nanoparticles and proteins. The samples appear clear to partially turbid.
III Specifications & Definitions
III.1 Quantitative Specifications
Requirements of this kind can be registered as follows:
- Throughput
- Precision
- Accuracy
- Resolution
- Reproducibility
- Electrophoretic mobility range
As each technique covers a similar ζ possible range (-100 mV to +100 mV), it is not possible to determine this requirement. However, electrophoretic mobility is different. In Equation 2, a nonpolar liquid (ε small) and/or in a viscous liquid (η large) can be seen; although the ζ potential could be significant, the electrophoretic mobility could be slight. This causes a small electrophoretic velocity if the electric field is not set extremely high, which is not preferable and could result in detrimental secondary outcomes.
It is better to employ a sensitive detection method such as PALS. In water at room temperature, one mobility unit (10-4 cm2 /V•s) corresponds to 12.8 mV. The classical methods are restricted to a few millivolts, as is ELS. But PALS can measure as low as 0.001 mobility units.
In water at room temperature, this would total 0.0128 mV, unfeasible to assess employing ELS. But in oleic acid, with a ratio of dielectric constant to viscosity at room temperature 1,000 times less than in water, it would total 12.8 mV, which would be simple to measure with PALS. See Table I shows some examples of this.
Table I. Mobility Ratios, ε/η normalized to water. Source: Brookhaven Instrument Corporation
| test |
| Water |
0.89 cP |
78 |
1.0 |
| Toluene |
0.56 cP |
2.38 |
0.05 |
| Ethylene Glycol |
17 cP |
40 |
0.03 |
| Oleic Acid |
26 cP |
2.46 |
0.001 |
Throughput is the total over the following times:
- Analysis time
- Cleanup time
- Sample preparation time
- Data reduction/printout/interpretation time
Sample preparation could be as small as a few minutes or need overnight wetting using a roller or orbital shaker. The measurement or analysis time is usually fairly short using the more recent instruments; however, for older, single particle methods, it may take longer. Statistical bias is feasible if not enough particles are recorded. This is not an issue employing the acoustic or light scattering techniques.
Similarly, data reduction and printout are rapid using modern computers. The duration to understand the data varies with each analyst and the set specifications. Cleanup time is frequently miscalculated as samples can get polluted with earlier ones if care is not employed in flow systems and in concentrates.
Disposable cells are useful in this instance. The number of measurements needed to be carried out every hour or every day should be identified. If a single point characterization (same solution conditions each time) is necessary or for the IEP, isoelectric point where the zeta potential is zero at a specific pH or additive strength? This type of measurement needs either an auto titrator or manual measurements following the alterations to the pH or the additive concentration.
III.2 Definitions
Precision is a means of identifying the proximity of an experimental outcome to the ‘true’ value. For methods that cannot be calibrated, or for any additional set of circumstances where a ‘true’ value is either undetermined or not well specified, then precision does not have meaning. Despite this, where more than one method is relevant, by contrast across multiple methods, various instruments and institutions, precision can be formed.
The NIST Standard Reference Material SRM 1980 consists of goethite at 500 mg/L, prepared under extremely strict phosphate and sodium perchlorate solution environments and at pH 2.5. Under these conditions the electrophoretic mobility, µep = (+2.53 ± 0.12) µ•cm/V•s with 95% confidence level. That is nearly +/-5%. Contrasted with particle sizing of spheres, electrophoretic mobility measurements are just fairly precise broadly.
Accuracy is an indication of the disparity in repeated measurements employing identical environments, including instrument, sample and operator. Accuracy (linked to systematic inaccuracy) and precision (linked with random inaccuracy) are linked: The outcomes of multiple measurements could band closely together (high precision, low random error), but the mean of the group could be a great distance from the true value (low accuracy, high systematic error).
Despite this, if a measurement is extremely exact, then repeated measurements will have gathered in proximity to the actual value. Nevertheless, exact mean values could comprise of either high or low precision. In such instances, precision restricts accuracy. Precision for many zeta potential measurements does not exceed 5%, reliant on the conductivity. At elevated conductivity (for instance, larger than 100 mM ionic strength), thermal impacts lead to lessened precision.
Resolution is a gauge of the lowest detectable discrepancies across different elements in a distribution. It is uncommon to discover a wide or multimodal zeta potential distribution and as such, resolution is not a key worry in zeta potential determinations. Combinations are the exception.
Reproducibility is a quantifier of the disparity across various machines, operators, sample preparations, etc. It becomes highly crucial when contrasting the findings generated on two separate machines of theoretically the same kind. A situation like this is fairly ordinary in the industry where many machines from one manufacturer and one model have been bought for employment in distinct labs and/or locations across the globe.
It is remarkable how frequently the resolution, conveyed as an array of values, surpasses the standard precision for just one of the machines. In these instances, it is beneficial to conduct round-robin tests on the same sample and employing the same set of specified conditions to separate any machine-to-machine variants.
III.3 Qualitative Specifications
Additional to quantitative requirements, there are qualitative requirements that are critical for the buying of any analytical instrument. These consist of the following:
- Support: Is there assistance available for training, service and throughout the course of the installation, within the warranty time and for the duration of the instrument's serviceable time? Instruments are now accessible from multiple parts of the world at half price, but they do not contain the kind of assistance most users anticipate.
- Ease-of-Use: Will the instrument be employed by experts who have been trained in how to use it or by users who lack experience and will only be using it temporarily? Despite the objective of a ‘one button’ device being commendable, it is seldom attained if for no other cause than sampling and sample preparation is not always one-button amenable.
- Versatility: Does the instrument manage aqueous samples in small, medium and large salt strengths? Can the instrument be employed in polar in addition to nonpolar liquids? Can the instrument be used with dilute samples or concentrates or both?
- Life-Cycle Cost: The instrument expense is just one element to think about. If labor intensive, the life cycle expense can be fairly large. Are cleanup expenses needed? The life-cycle cost may be higher than different options.
Of each of these matters, support is probably the most critical. When deciding between sellers of similar or the same equipment, the one which offers better support may win overall. Customers frequently presume the biggest vendor or the one with the most impressive website offers the best support.
Frequently, these are the sellers with the biggest overheads who may not realize that the sales of analytical instruments for zeta potential determination generate plenty of profits to stay the course. Over the past 25 years, the leaders in zeta potential instrumentation were smaller, committed companies.
IV. Narrow Down the Possibilities and Make a Choice
The question must be posed about how much information is required from the measurement. Is just the sign of the zeta potential necessary or the monitoring of changes with time and additive? Is it a specific requirement to locate the IEP, the isoelectric point, the pH, or intensity of the additive that leaves the zeta potential zero? Is it necessary to automate that with a devoted auto titrator whose operation is incorporated with that of the central instrument?
If the addition of particle sizing and/or molecular weight is intended, the ELS and PALS are the methods of choice. If cost is the key factor, the classical optical selections are usually, but not always, the cheapest if labor costs are not taken into account. They are usually slower and affected by operator bias more than alternative methods. The acoustic instruments are often the costliest and the light scattering choices are somewhat of a middle ground.
If it is necessary to take measurements without dilution, the acoustic instruments are the most appropriate. Cleanup and cross contamination are a matter of concern and additionally, they are usually considerably more difficult to use. It is pertinent to look at the appendix before making preliminary measurements made for useful suggestions.
Appendix: Sampling and Sample Preparation
Once the selection has been narrowed to one or two methods and one to three sellers in each group. Following this, samples should be sent to each and results should be requested. By evaluating the outcomes, the answer to the question about mobility range should be identified. It is also possible to determine the kind of assistance the company provides from the timeliness of the vendor's queries and outcomes. The vendor that poses plenty of relevant queries regarding the sample properties is likely to be the best at providing future support.
To establish the electrophoretic mobility range of interest, preliminary measurements should be taken, with particular care to guarantee the sampling and sample preparation are identical for every sample sent out for analysis. Primarily, if the sample is not a frequently occurring one, it should be assumed that there might be more than one cycle of sampling, sample preparation and range establishment.
For frequently occurring samples, references should be consulted. It is easy to think that an instrument with the biggest, claimed range of use is the most effective. Although it may be appropriate for the sample at hand, it might not be the best selection.
The initial issue is to make sure the sample is typical of the whole and is split without bias across the numerous samples to be sent. There is a large literature on sampling it would be useful to make reference to, but to put it succinctly: If the sample is a dry powder, it is a necessary sample from a moving stream of dry particles and definitely across the entire stream to prevent segregation by size.
Once the sample is lowered to a manageable size (around one kilogram or less), a spinning riffler should be employed to create smaller, equal portions that are evenly dispersed with regard to size distribution. (This is increasingly critical for particle sizing, less essential for zeta potential.) If the sample is a suspension in a liquid, it should be gently combined and meticulously employing a roller (larger sizes) or an orbital shaker (colloidal and nanoparticle dispersions) prior to splitting into smaller lots is made.
For big, dense particles, specific care should be given to segregation by sedimentation throughout the separation process. The second issue is to guarantee that all of the vendors are provided identical instructions for sample preparation. If not, the assortment of results gathered will be more indicative of the various sample preparations than the different instrumental methods or the different vendors.
This is particularly true if different wetting, dispersing, or pH agents are employed, as these will alter the surface charge density. Literature should be consulted as well as vendors for their advice on how to prepare the sample correctly is not known.
The following are the questions any decent vendor should ask and the information that should be given to them.
- Are the samples powders, dilute dispersions, dissolved polymers, or proteins, or concentrates?
- If concentrates, are they dilutable without altering the zeta potential? If they are, what is the diluent?
- What additives, if any, are needed? Additives that preferentially adsorb onto the surface will impact zeta potential.
- Wetting agents reduce the contact angle across particles and air, permitting liquid to wet the surface.
- Dispersing agents add repulsive forces across particles with ample length to take zeta potential measurements.
- Stabilizing agents add long-term repulsive forces between particles and are mainly employed for long-term product performance.
- NOTE: Any single additive can offer two but not all three of these properties. It is also worth reiterating: Additives (wetting, dispersing, stabilizing agents) going to the surface change the zeta potential.
- How much energy, if any and for what length of time is it necessary to disperse the particles? If a new surface is formed, the zeta potential will more than likely be altered.
- Minimal energy is best, like manual swirling, gentle rolling or orbital shaking. A low power ultrasonic bath is better than a strong sonic probe that may produce new surface area and alter the size distribution and zeta potential. It varies on the sample.
To correctly understand the measurements, one or more of the following bits of information regarding the particle and liquid properties is either necessary or helpful in making estimates:
Particle Properties: Most critical are any surface treatments
- Type: latex, clay, organic pigment, drugs, oil/water emulsion, etc.
- Knowledgeable analysts can estimate numerous particle properties simply by possessing the knowledge of the kind of chemistry they are dealing with.
- Surface Treatments: The main element of zeta potential before the addition of wetting and dispersing agents.
Medium Properties
- Type: water or chemical name of organic solvent
- Viscosity, ho and temperature are necessary in calculations.
- Dielectric constant (relative permittivity), eo and the temperature are necessary in calculations.
- pH of aqueous suspensions can impact zeta potential of electrostatically stabilized particles, so it is useful to know what the starting point is prior to additional steps taken.
- Ionic Strength, I, is related to the conductivity of an aqueous-based sample and is a function of the concentration of free ions like salts, acids/basis and ionic additives such as wetting and dispersing agents.
References
i Small here means mostly below one micron: colloids, nanoparticles, proteins, etc. While all macroscopic surfaces when wetted may
develop charge, and while such a charge density may have a profound influence on what adheres to that surface, it is the zeta potential
of small particles that determines dispersion stability.
ii The permittivity of free space, εo = 8.854 x 10-12 Farad/m.
iii D. Fairhurst, American Pharmaceutical Review, vol. 16, April, 2013.

This information has been sourced, reviewed and adapted from materials provided by Brookhaven Instrument Corporation.
For more information on this source, please visit Brookhaven Instrument Corporation.