Nov 14 2013
For structural biologists, the first step in determining a protein's precise molecular structure is often the hardest: coaxing the protein to grow into the orderly, three-dimensional crystals that are the starting material for most structural studies. For particularly difficult cases, it can take years to generate usable crystals – and sometimes the protein never crystallizes despite intense effort. Howard Hughes Medical Institute (HHMI) scientists have developed a new method that generates a high-resolution protein structure from crystals one-million times smaller than those needed for X-ray crystallography, the most common method for determining protein structure.
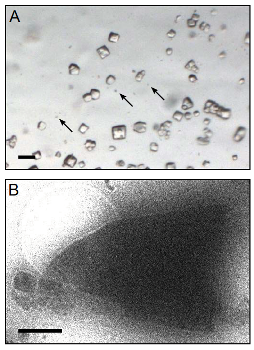 mages of lysozyme microrystals. (A) Light micrograph showing 524 lysozyme microcrystals (three examples indicated by arrows) in comparison with 525 larger crystals of the size normally used for X-ray crystallography. Scale bar is 50 526 ìm. (B) Lysozyme microcrystals visualized in over-focused diffraction mode on the 527 cryo-EM prior to data collection. The length and width of the crystals varied from 2 528 to 6 ìm with an estimated thickness of ~0.5 to 1 ìm. Scale bar is 1 ìm.(Tamir Gonen, Janelia Farm Research Campus/HHMI)
mages of lysozyme microrystals. (A) Light micrograph showing 524 lysozyme microcrystals (three examples indicated by arrows) in comparison with 525 larger crystals of the size normally used for X-ray crystallography. Scale bar is 50 526 ìm. (B) Lysozyme microcrystals visualized in over-focused diffraction mode on the 527 cryo-EM prior to data collection. The length and width of the crystals varied from 2 528 to 6 ìm with an estimated thickness of ~0.5 to 1 ìm. Scale bar is 1 ìm.(Tamir Gonen, Janelia Farm Research Campus/HHMI)
The new technique, called MicroED, has the potential to accelerate structural biologists' efforts and to expand the repertoire of proteins whose high-resolution structures can be solved. “Biochemically, it is always easier to generate smaller crystals,” says structural biologist Tamir Gonen, who developed the technique with colleagues in his lab at HHMI's Janelia Farm Research Campus. “There are many proteins where you either don't get crystals or you get crystals that are very, very small. They might be good enough for microED.”
Gonen and colleagues describe MicroED in a report published November 19, 2013, in the open access journal eLife. Using Gonen’s new technique, scientists can use electrons from an electron microscope to determine the structure of protein microcrystals. This approach, known as electron crystallography, had previously been limited to studies of proteins that could be grown into very thin, two-dimensional crystals. “People have put 3D crystals in electron microscopes before, but no one was able to solve their structures,” Gonen says.
That's because the radiation from the microscope's high energy electron beam – which is scattered by the protein crystal, creating diffraction patterns that researchers use to deduce structure -- rapidly destroys protein crystals. The crystals only survive two or three exposures to a “low-dose” beam, Gonen explains. Yet many more diffraction patterns are needed to reveal a protein’s structure.
That means to generate enough data to generate a structure, researchers must collect and analyze the diffraction patterns from thousands of different crystals. Combining that data is difficult even for two-dimensional crystals, but for three-dimensional crystals, it has proven an insurmountable task. “If the data comes out of thousands of different crystals, you have no idea where you are in reciprocal space – no idea how these various diffraction patterns relate to each other. There is no way to index everything and determine the structure,” Gonen says.
So Gonen, microscopy specialist Dan Shi, and their colleagues decided to simplify the data processing by collecting many diffraction patterns from a single crystal. “When we collect multiple diffraction patterns from a single crystal, they have continuity between them. We know exactly how they relate,” he explains.
“We didn't know if we could actually collect an entire data set out of a single crystal, but we decided to try,” he says. The team knew that to preserve the crystal for repeated exposures, they would have to dramatically reduce the electron dose from the microscope. “Most people use the electron microscope for imaging,” Gonen explains. “But since we only wanted to collect diffraction patterns, we were able to use about 200 times less than what is usually considered a low dose.”
To test their idea, Brent Nannenga, a postdoctoral researcher in Gonen's lab, grew very small crystals of a protein called lysozyme, which has a known structure. They applied these microcrystals, which were so small they appeared as mere specks under a light microscope, to a small carbon grid. After placing the grid into the microscope, they programmed the instrument so that it would rotate the grid one degree at a time, collecting a diffraction pattern after each rotation. Ninety-degrees worth of data were collected.
Matt Iadanza, a graduate student in Gonen’s lab, developed software to index and integrate the 90 diffraction patterns, enabling the team to generate a complete structure of the lysozyme protein. To improve the quality of the structure, they repeated this process for two more crystals – each lying in a different orientation on their grid – then combined the data into a single structure.
That structure was consistent with the known structure of lysozyme, with a resolution of 2.9 angstroms. “That's exactly where you want to be for biological proteins,” Gonen says, explaining that this level of detail elucidates many of the structural motifs that underlie protein function. “At that resolution, you really can begin to understand molecular mechanism,” he says. By further refining their data processing programs, he adds, his team should be able to improve the resolution that MicroED can provide. Data was already collected to 1.7 angstrom resolution so it’s only a matter of time.
Gonen's team is now working with Andrew Leslie at the Medical Research Council Laboratory of Molecular Biology to integrate MicroED data processing into standard X-ray crystallography software (MOSFLM), to make the technique more accessible to other laboratories. In the meantime, he says, many labs have brought their protein microcrystals to Janelia, and the team is investigating the technique's potential to solve unknown structures.
“There's a lot of excitement,” Gonen says, “because lots of people have microcrystals and they don't even know it. What we've found is that a crystallization drop that has large crystals in it always has microcrystals. And even if you can't see crystals under a light microscope, it doesn't mean that you don't have microcrystals.”
Press release avilable from http://www.newswise.com/