Mar 30 2016
A new 3-D modeling and data-extraction technique is about to transform the field of X-ray crystallography, with potential benefits for both the pharmaceutical industry and structural biology.
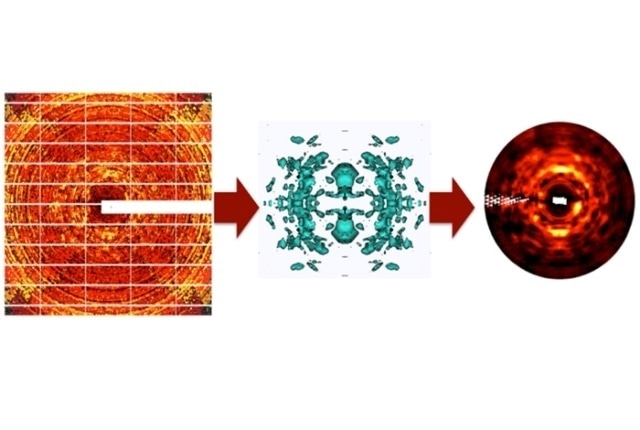 Conventional protein X-ray diffraction images are processed to remove the sharp Bragg reflections, producing noisy images of diffuse intensity (left). Images from multiple crystal orientations are integrated into a 3-D dataset in which the signal is statistically averaged (center, showing a level surface of diffuse intensity in three-dimensions). The 3-D data show greatly enhanced diffuse features, as seen in simulated diffraction images obtained using the integrated data (right, compare to left panel). The 3-D data are used to validate and refine models of protein motions.
Conventional protein X-ray diffraction images are processed to remove the sharp Bragg reflections, producing noisy images of diffuse intensity (left). Images from multiple crystal orientations are integrated into a 3-D dataset in which the signal is statistically averaged (center, showing a level surface of diffuse intensity in three-dimensions). The 3-D data show greatly enhanced diffuse features, as seen in simulated diffraction images obtained using the integrated data (right, compare to left panel). The 3-D data are used to validate and refine models of protein motions.
A paper this week in Proceedings of the National Academy of Sciences describes the improved blending of experimentation and computer modeling, extracting valuable information from diffuse, previously discarded data.
“The accomplishment here is to demonstrate that we can analyze conventionally collected protein crystallography data and pull out background features that are usually thrown away,” said Michael E. Wall, a scientist in the Computer, Computational, and Statistical Sciences Division at Los Alamos National Laboratory and co-corresponding author of the paper with James S. Fraser, Assistant Professor in the Department of Bioengineering and Therapeutic Sciences at University of California, San Francisco.
“What’s been reclaimed is information about how the protein moves, the more detailed dynamics within the sample,” Wall said. Traditional crystallography data provide a blurred picture of how the protein moves, like overlaying the frames of a movie. Our approach sharpens the picture, he noted, providing information about which atoms are moving in a concerted way, such as ones on the swinging arm of the protein or on opposite sides of a hinge opening or closing, and which ones are moving more independently.
“This is a method that will eventually change the way X-ray crystallography is done, bringing in this additional data stream in addition to the sharply peaked Bragg scattering, which is the traditional analysis method,” Wall said We’re working toward using both data sets simultaneously to increase the clarity of the crystallography model and more clearly map how proteins are moving.”
In the work described in the paper, the 3-D diffuse scattering data were measured from crystals of the enzymes cyclophilin A (a type of protein) and trypsin (an enzyme that acts to degrade protein) at Stanford Synchrotron Radiation Lightsource (SSRL), a U.S. Department of Energy (DOE) Office of Science User Facility. The measurements were extracted and movements were modeled using computers at Los Alamos National Laboratory, Lawrence Berkeley National Laboratory, and the University of California, San Francisco. The ongoing computational work includes simulations on Conejo and Mustang, supercomputing clusters in Los Alamos National Laboratory’s Institutional Computing Program.
Averaging relatively weak features in the data improves the clarity of the imaging of diffuse features, which has value as researchers have had an increasing interest in the role of protein motions. In designing a new drug, for example, one seeks to produce a small molecule that binds to a functional site on a specific protein and blocks its activity. With better modeling, adapted to more closely match experimental diffuse data, the steps toward a new pharmaceutical product, can be reduced, by more accurately accounting for protein motions in drug interactions.
The new approach can improve new and ongoing experiments and could potentially be used to explore data from previously conducted crystallography experiments if the level of background noise is not too severe.
“Data coming off modern X-ray sources with the latest detectors are tending to be the type of data we can best analyze,” Wall said, but “some of the older data could be reexamined as well.”
With this new method, scientists can experimentally validate predictions of detailed models of protein motions, such as computationally expensive all-atom molecular dynamics simulations, and less expensive “normal mode analysis,” in which the protein motions are modeled as vibrations in a network of atoms interconnected by soft springs.
A key finding is that normal modes models of both cyclophilin A and trypsin resemble the diffuse data; this creates an avenue for adjusting detailed models of protein motion to better agree with the data. “We are planning to add in future a refinement step to increase accuracy even more,” Wall said. A more detailed model provides a more direct connection between protein structure and biological mechanisms, which is desired for pharmaceutical applications.
This use of diffuse scattering data illustrates the potential to increase understanding of protein structure variations in any X-ray crystallography experiment. Said Wall, “This represents a significant step toward moving diffuse scattering analysis into the mainstream of structural biology.”
The paper: “Measuring and modeling diffuse scattering in protein X-ray crystallography,” by authors Andrew H. Van Benschoten, Lin Liu (both of the Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco), Ana Gonzalez (Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory), Aaron S. Brewster, Nicholas K. Sauter, (both of Molecular Biophysics and Integrated Bioimaging Division, Lawrence Berkeley National Laboratory) James S. Fraser (Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco) and Michael E. Wall (Los Alamos National Laboratory)
Funding: This work was supported by the University of California, Office of the President, Multicampus Research Programs and Initiatives Grant MR-15-338599, and the Program for Breakthrough Biomedical Research, which is partially funded by the Sandler Foundation. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the US Department of Energy, Office of Basic Energy Sciences. The Stanford Synchrotron Radiation Lightsource Structural Molecular Biology Program is supported by the US Department of Energy Office of Biological and Environmental Research, and by the NIH, National Institute of General Medical Sciences. Ongoing computer simulations are supported by the Los Alamos National Laboratory Institutional Computing Program. Sauter was supported by NIH Grant GM095887. Fraser was supported by a Searle Scholar Award from the Kinship Foundation, a Pew Scholar Award from the Pew Charitable Trusts, a Packard Fellowship from the David and Lucile Packard Foundation, NIH Grant OD009180, NIH Grant GM110580, and National Science Foundation Grant STC-1231306. Wall was supported by the US Department of Energy through the Laboratory-Directed Research and Development Program at Los Alamos National Laboratory.
Image caption: Conventional protein X-ray diffraction images are processed to remove the sharp Bragg reflections, producing noisy images of diffuse intensity (left). Images from multiple crystal orientations are integrated into a 3-D dataset in which the signal is statistically averaged (center, showing a level surface of diffuse intensity in three-dimensions). The 3-D data show greatly enhanced diffuse features, as seen in simulated diffraction images obtained using the integrated data (right, compare to left panel). The 3-D data are used to validate and refine models of protein motions. (Image courtesy Los Alamos National Laboratory)