Solid-state nuclear magnetic resonance (NMR) spectroscopy — a method that calculates the frequencies produced by the nuclei of certain atoms exposed to radio waves in a powerful magnetic field — can be used to establish chemical and 3D structures as well as the dynamics of materials and molecules.
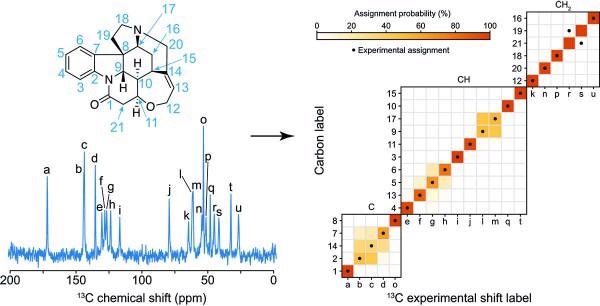 Probabilistic assignment of the 13C NMR spectrum of crystalline strychnine. (Image Credit: NCCR MARVEL).
Probabilistic assignment of the 13C NMR spectrum of crystalline strychnine. (Image Credit: NCCR MARVEL).
An essential initial step in the study is, however, the so-called chemical shift assignment. This includes allocating each peak in the NMR spectrum to a specific atom in the molecule or material under study. This can be a predominantly complex task.
Assigning chemical shifts experimentally can be tough and usually needs laborious multi-dimensional correlation experiments. Assignment by comparison to statistical analysis of experimental chemical shift databases would be an alternative answer, but no such database for molecular solids exists.
A group of scientists, including EPFL professors Lyndon Emsley, head of the Laboratory of Magnetic Resonance, Michele Ceriotti, head of the Laboratory of Computational Science and Modelling, and PhD student Manuel Cordova, decided to resolve this issue by formulating a technique of assigning NMR spectra of organic crystals probabilistically, straight from their 2D chemical structures.
They began by developing their own database of chemical shifts for organic solids by integrating the Cambridge Structural Database (CSD), a database of over 200,000 three-dimensional organic structures, with ShiftML, a machine learning algorithm they had formulated together earlier that facilitates the prediction of chemical shifts straight from the structure of molecular solids.
At first, illustrated in a Nature Communications article in 2018, ShiftML uses DFT calculations for training, but can then carry out precise predictions on new structures without executing extra quantum calculations.
Though DFT accuracy is accomplished, the technique can measure chemical shifts for structures with ~100 atoms in seconds, decreasing the computational cost by a factor of nearly 10,000 compared to existing DFT chemical shift calculations.
The accuracy of the technique does not rely on the size of the structure studied and the prediction time is linear in the number of atoms. This paves the way for calculating chemical shifts in circumstances where it would have been unachievable before.
In the Science Advances article, they used ShiftML to estimate shifts on over 200,000 compounds derived from the CSD and then related the shifts attained to topological representations of the molecular environments.
This involved building a graph signifying the covalent bonds between the atoms in the molecule, spreading a specific number of bonds away from the central atoms. They then compiled all the identical cases of the graph in the database, allowing them to attain statistical distributions of chemical shifts for each motif.
The representation is an interpretation of the covalent bonds around the atom in a molecule and does not comprise any 3D structural features: this enabled them to acquire the probabilistic assignment of the NMR spectra of organic crystals straight from their two-dimensional (2D) chemical structures via a marginalization scheme that integrated the distributions from all the atoms in the molecule.
After building the chemical shift database, the researchers aimed to calculate the assignments on a model system and applied the method to a set of organic molecules for which the carbon chemical shift assignment has already, or at least partly, been established experimentally: lisinopril, theophylline, cocaine, strychnine, thymol, AZD5718, ritonavir and the K salt of penicillin G.
The assignment probabilities acquired straight from the 2D representation of the molecules were found to suit the experimentally established assignment in a majority of cases.
Finally, they assessed the performance of the framework on a benchmark set of 100 crystal structures with between 10 and 20 dissimilar carbon atoms. They used the ShiftML predicted shifts for each atom as the precise assignment and omitted them from the statistical distributions used to assign the molecules. The precise assignment was discovered among the two most likely assignments in over 80% of cases.
This method could significantly accelerate the study of materials by NMR by streamlining one of the essential first steps of these studies.
Manuel Cordova, PhD Student, EPFL
Journal Reference:
Cordova, M., et al. (2021) Bayesian Probabilistic Assignment of Chemical Shifts in Organic Solids. Science Advances. doi.org/10.1126/sciadv.abk2341.