Reviewed by Alex SmithJun 16 2022
The use of theoretical calculations to uncover the thermodynamics and kinetics of catalysis has become indispensible.
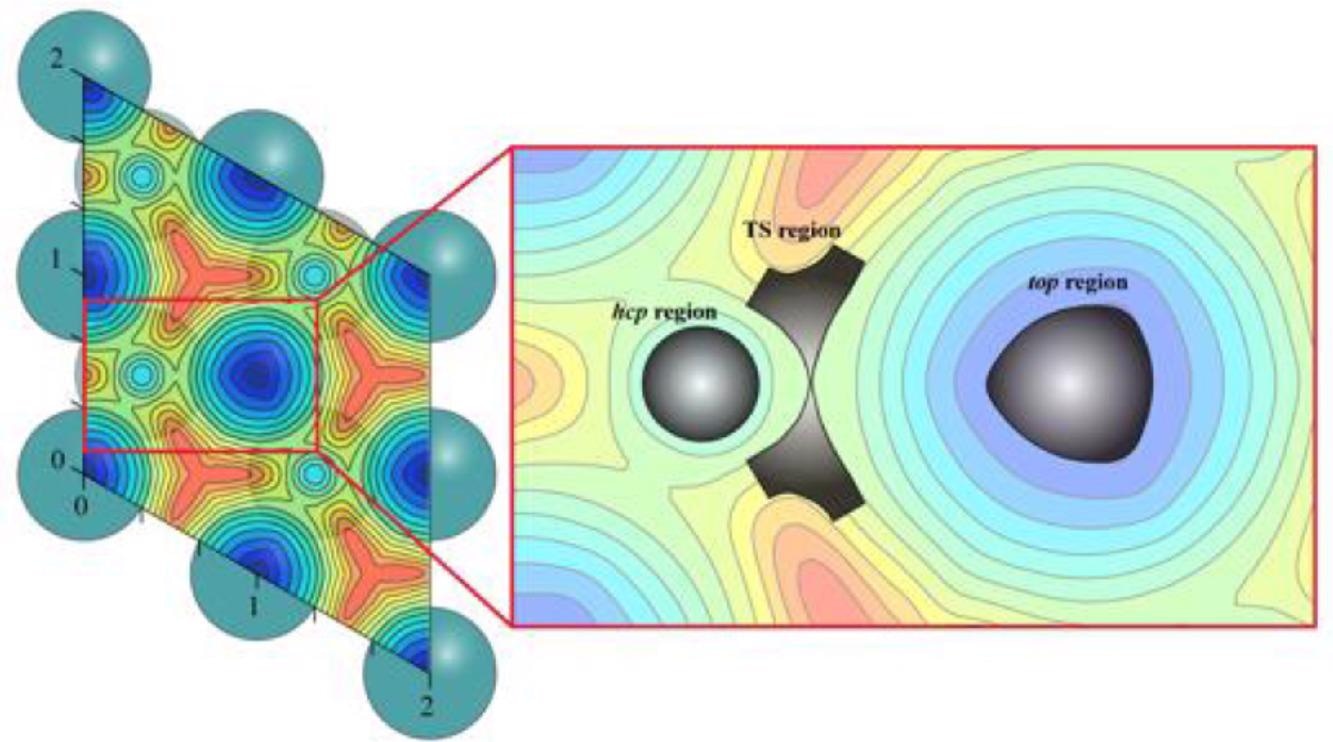 Illustration of the Research. Image Credit: Prof. Zhuang’s group
Illustration of the Research. Image Credit: Prof. Zhuang’s group
The most common technique in theoretical catalysis is the static computational strategy, in which the reaction thermodynamics and kinetics are calculated using a few fixed geometries at zero temperature and certain ideal statistic mechanics models.
As a result, the compliance between ideal models and reality situations limits the accuracy of the frequently employed static technique. Ab initio molecular dynamics (AIMD) simulation, on the other hand, is a tried-and-true method for going beyond static stationary states.
However, due to the high computing cost of extended time scale simulations, obtaining convergent thermodynamics and kinetics is extremely challenging.
Prof. Jun Chen and Prof. Zhening Chen from Prof. Wei Zhuang’s group at the Chinese Academy of Sciences’ Fujian Institute of Research on the Structure of Matter recommended a highly efficient dynamic computational strategy for the calculation of thermodynamics and kinetics in heterogeneous catalysis based on a combination of efficient potential energy surface (PES) and molecular dynamics (MD) simulations in a recent study published in Chinese Chemical Letters.
The researchers obtained a long-timescale MD at the microsecond level using chemisorbed CO on Ru (0001) surface as the illustrative model catalytic system, and the neural network-fitted PES showed high efficiency.
They were able to obtain reliable temperature-dependent thermodynamics and kinetics at seven varying temperatures from 300 to 900 K, which goes beyond the popular static approach and the transition state theory, supplying a more detailed version of catalytic processes under realistic conditions.
The researchers also said that a dynamic computational technique based on efficient PES and MD simulations is easily available as an accurate but efficient method for evaluating the exact thermodynamics and kinetics of catalytic processes under realistic settings.
The findings can be utilized as a baseline for further research into dynamic heterogeneous catalysis methods.
This research demonstrates the efficiency and reliability of a dynamic computational strategy that focuses on efficient PES and MD simulations, which is expected to be a powerful tool for statistical thermodynamics and kinetics calculations in catalysis when used in conjunction with appropriate enhanced sampling approaches.
Journal Reference:
Chen, J., et al. (2022) Toward accurate and efficient dynamic computational strategy for heterogeneous catalysis: Temperature-dependent thermodynamics and kinetics for the chemisorbed on-surface CO. Chinese Chemical Letters. doi:10.1016/j.cclet.2022.03.080.