Aug 25 2016
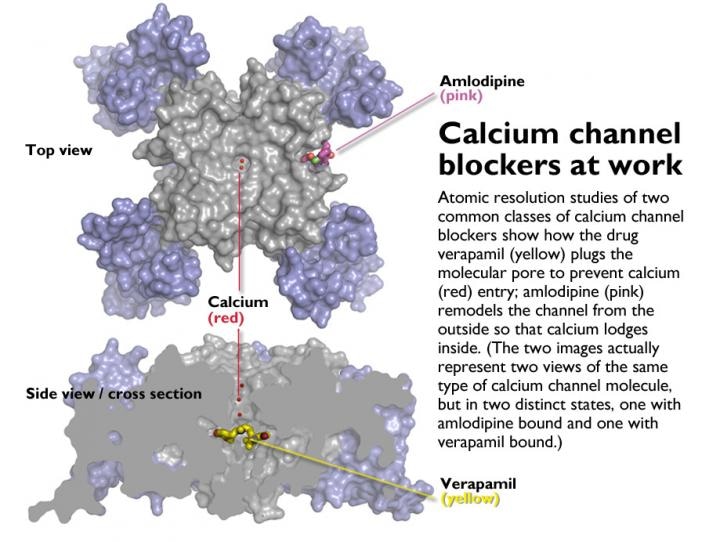 Atomic resolution studies of two common calcium channel blockers, one that treats irregular heart beats, and another that controls high blood pressure and angina. Credit: Catterall and Zheng Labs/University of Washington
Atomic resolution studies of two common calcium channel blockers, one that treats irregular heart beats, and another that controls high blood pressure and angina. Credit: Catterall and Zheng Labs/University of Washington
Two specific classes of calcium channel blockers, commonly prescribed for patients with heart diseases, develop separate therapeutic effects based on their actions at varied sites on the calcium channel molecule. This discovery was made through an atomic level analysis.
Patients from all over the world control cardiovascular problems by taking calcium channel blockers.
The latest issue of the advanced online edition of Nature features a study in which researchers explain how the basic mode of action of two distinctive chemical classes of these drugs differs. The knowledge was obtained by examining the atomic structure of their binding sites.
The project was headed by UW Medicine researchers Dr. William A. Catterall, University of Washington professor and chair of pharmacology, and Ning Zheng, UW professor of pharmacology and investigator of the Howard Hughes Medical Institute. Lin Tang, UW postdoctoral research scientist in pharmacology, was the first author on the Nature report.
Calcium channel blockers were first detected and approved as medications in the previous half-century. These blockers have since emerged as significant therapies ideal for cardiovascular disorders.
The recent findings could inform the design of improved, safer versions of calcium channel blockers for controlling high blood pressure, chest pain, and heart beat irregularities.
Calcium channel blockers that are used for treating heart arrhythmias, such as verapamil, were compared to those used for hypertension or angina, such as amlodipine.
Diltiazem and a third class of calcium channel blockers reduce the heart rate and dilate the blood. Those drugs were not considered in this study.
The researchers focused on studying how calcium channel blocker molecules work together with calcium channels, the molecular pores that control the travel of calcium ions through a cell membrane.
The opening of these pores in heart muscle cells or in the smooth muscle cells in veins and arteries results in calcium rushing into the cells and prompting the heart to pump blood and for the arteries to contract and narrow their diameter, increasing blood pressure.
Interfering with these molecular pores, allows calcium channel blockers to subdue a majorly powerful cardiovascular response capable of causing high blood pressure or an irregular heart beat.
Pharmacologists believed that verapamil-like drugs were capable of physically blocking calcium channel pores in order to prevent the entry of calcium into the cell, restoring a normal heart rhythm. However, amlodipine-like drugs - also known as dihydropyridines - were assumed to indirectly prevent calcium channel activation and pore opening, resulting in the prevention of angina and high blood pressure.
However, particular structures behind these varied mechanisms of action continued to be uncharted.
X-ray crystallography helps to determine the arrangement of atoms within a huge protein molecule. The progress in X-ray crystallography and in the functional analysis of ion channels is currently enabling researchers to investigate the submolecular depths of the actions of the drugs.
Researchers observed where the drug molecules bound to calcium channels, and also how this integration alters the functioning of the channels.
The binding spot of the angina medication, amlodipine, and the blood pressure, was identified to be the outside edge of the calcium channel molecule. Voltage sensors that are present around the central pore are sensitive to electrical potential.
The binding site is arranged on the central pore structure’s outside edge between two of the four subunits of the calcium channel molecule. The binding closes the channel by altering its shape and permanently placing a calcium ion within it.
The amlodipine subtly remodels the pore so that the calcium ion is pulled to one side and just sticks there the whole time, as if it were locked up.
Ning Zheng, Professor, University of Washington
In contrast, the verapamil molecule has the potential to plug a calcium channel’s central cavity and directly blocks the calcium ion-conducting pathway.
During palpitation of the heart, verapamil takes advantage of the recurrent openings of the calcium channels, just as its does during atrial flutter or atrial fibrillation. Frequent opening of the pore increases the chances of the verapamil molecule slipping into the central cavity and sealing off the pore.
Verapamil appears to bind better to calcium channels in the rapidly beating parts of the heart and slows them down.
Dr. William A. Catterall, Researcher, University of Washington
He highlighted that Bertil Hille, his UW colleague and professor of physiology and biophysics, along with his associates previously illustrated the effects of rapid firing frequency on sodium channel blockade in their studies dealing with local anesthetic drugs such as lidocaine that is used for preventing pain in surgery and dentistry.
Calcium channels present in the blood vessel cells are generally closed in their resting state. Amlodipine molecules adjust the voltage-dependent activation of calcium channels, and do not rely on recurrent openings of the channel in order to enter the pore.
This is the reason why amlodipine-like drugs, which help relaxing the blood vessels, are capable of treating specific causes of high blood pressure and the squeezing, tight pain of angina without major effects on the heart itself. This distinguishes them from the verapamil-like drugs, which support calcium channels in cells active in the heart’s electrical circuitry.
Calcium channel blockers that accurately fit in place can be developed by understanding details related to the two binding sites. A much more accurate shape could also prevent next-generation versions of the blockers from unintentionally positioning themselves with the wrong binding sites thus resulting in unnecessary side effects.
“Calcium channel blockers are relatively safe drugs,” Catterall explained, “but toxicity can arise from overdoses that can lead to ventricular arrhythmias or to too strong depression of the contraction of the heart or smooth muscle cells.”
He went on to say that structure-based, enhanced drug design might make room for small and efficient drug doses that are a lot safer and specific. Altering the drug design could prevent another possible contributor to unnecessary side effects, for example.the off-target blocking of sodium channels by calcium channel blockers.
The new research was based on bacterial ion channels, which are known to be the ancestors of calcium and sodium channels in other life forms, including humans, fish, flies and worms.
These ancient channels in bacteria still recognize the drugs designed for people. The experiments done by our second author Tamer Gamal El-Din, acting assistant professor of pharmacology, showed that these drugs act in the same way on bacterial channels as they do on those in mammals. It’s remarkable that the most basic bacterial channels respond to these modern medicines for treating arrhythmias and certain other cardiovascular diseases.
Dr. William A. Catterall, Researcher, University of Washington
The research received financial aid from the federally sponsored Advanced Light Source at the Lawrence Berkeley National Laboratory, where the tiny structures of the unbound and bound calcium channels could be examined.
Interestingly, Zheng explained that the experiments performed at the synchrotron beam line in California could be handled long-distance from the UW in Seattle through a simple laptop computer operated by Lin Tang and other investigators involved in this research.
Other researchers on this project were Teresa M. Swanson, UW pharmacology Ph.D. candidate; David C. Pryde, Pfizer Research Unit at Worldwide Medicinal Chemistry in the United Kingdom; and Todd Scheuer, UW research professor of pharmacology.
The project featured in the latest issue of Nature was supported by the National Heart, Lung and Blood Institute of the National Institutes of Health (grant R01 HL112808), a National Research Service Award (grant T32GM008268) and the Howard Hughes Medical Institute.